
[ad_1]
L’innovation pharmaceutique sauve des vies. Mais tous les «nouveaux» médicaments sont vraiment nouveaux.
Les brevets sont conçus pour récompenser les inventions de percée en accordant aux inventeurs les droits de monopole temporaire pour récupérer les coûts de la recherche et du développement et pour encourager l’innovation future. Mais les entreprises peuvent également exploiter le système de manière à rendre les médicaments plus coûteux et moins accessibles aux patients. Une étude 2023 a révélé que 78% des médicaments associés à de nouveaux brevets n’étaient pas réellement de nouveaux médicaments mais des modifications mineures.
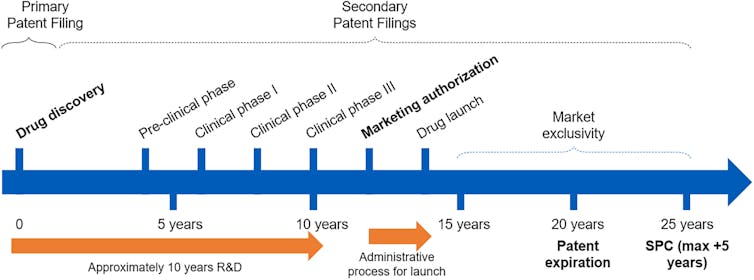
Après avoir obtenu le brevet principal d’un médicament, les sociétés pharmaceutiques en déposent souvent des droits de monopole supplémentaires. Cette pratique – appelée Evergreening – peut couvrir de nouvelles doses, des méthodes d’administration, des combinaisons de médicaments et des conditions. Bien que certains de ces brevets secondaires améliorent l’efficacité ou la commodité du traitement, beaucoup ont peu d’effet sur les résultats pour la santé. Plus souvent, ces changements ultérieurs sont principalement utilisés pour prolonger stratégiquement l’exclusivité du marché, retarder la concurrence des génériques et maintenir les prix des médicaments élevés.
Ces pratiques soulèvent des préoccupations concernant l’accès aux médicaments et l’abordabilité, en particulier lorsque les entreprises utilisent des ajustements mineurs pour bloquer les alternatives moins chères, avec peu d’avantages pour les patients. Pourtant, la distinction entre des améliorations vraiment innovantes et des extensions de faible valeur a été difficile pour les régulateurs et les tribunaux.
Je suis un économiste qui étudie l’innovation et la numérisation sur les marchés des soins de santé. Mon collègue Dennis Byrski et moi nous sommes concentrés sur la façon dont la transparence réglementaire joue un rôle dans la réduction des brevets faibles. Nos recherches récemment publiées ont révélé que lorsque les données des essais cliniques deviennent publiques, cette divulgation rend plus difficile pour les entreprises d’obtenir des brevets pour des changements progressifs qui ajoutent peu d’avantages thérapeutiques pour les patients.
Qu’est-ce qui rend un médicament brevetable?
Selon l’Organisation mondiale de la propriété intellectuelle, une invention brevetable doit être nouvelle et non évidente.
La nouveauté signifie que l’invention n’a pas été documentée auparavant dans des informations accessibles au public – telles que des brevets, des publications ou des produits – dans des domaines liés à une invention avant la date de dépôt. Ces informations sont souvent appelées arts antérieurs.
La non-évidence signifie que l’invention ne serait pas évidente – un ajustement ou une étape de routine facile dans le processus – pour une personne qualifiée dans le domaine sur la base des connaissances existantes. Par exemple, si le TAR antérieur révèle qu’une nouvelle thérapie combinée améliore les résultats du traitement, les responsables peuvent considérer les brevets ultérieurs en utilisant le même cocktail de médicament que évident et refuser d’accorder ou d’appliquer le brevet.
Pour les médicaments, ces deux concepts sont profondément liés à la sécurité et à l’efficacité. Si une entreprise reformule un médicament – par exemple, en modifiant un ingrédient inactif ou en peaufinant la dose – il n’est pas toujours facile de déterminer si ces changements améliorent la santé des patients sans plus de tests dans la clinique.
Selon les lignes directrices de l’Office européen des brevets, les résultats des essais cliniques peuvent être essentiels à l’art antérieur, en particulier lorsqu’ils révélaient des avantages thérapeutiques inattendus ou non divulgués. Les conseillers en brevets ont également noté que les preuves des essais peuvent jouer un rôle décisif dans l’évaluation de la nouveauté et de la non-évidence.
Cependant, les résultats complets des essais cliniques ne sont souvent pas disponibles ou non divulgués avant le début du processus d’autorisation de marketing, lorsqu’une entreprise soumet une demande complète aux régulateurs pour approuver officiellement un médicament à vendre.
En fait, alors que les régulateurs européens des médicaments encouragent fortement les entreprises à divulguer les données d’essai cliniques au début du processus, les entreprises peuvent reporter la publication des données d’étude jusqu’à sept ans après la fin de l’essai ou jusqu’à ce que le médicament soit sur le marché – selon la première éventualité. Ce dernier est plus contraignant pour les entreprises qui souhaitent retarder la libération de points de données critiques pour éviter la concurrence.
L’autorisation marketing modifie le jeu
Compte tenu du long processus de développement de médicaments, la plupart des entreprises déposent le brevet principal d’un médicament dès le début, souvent avant de commencer les essais cliniques et d’obtenir des données sur la sécurité et l’efficacité du traitement.
Ces informations sont requises lors de la demande d’autorisation de marketing et sont généralement divulguées grâce à des résultats détaillés de l’essai clinique de phase 3. Ces données peuvent ensuite devenir des TAR préalables pour évaluer les demandes de brevet ultérieures, ce qui rend plus difficile l’obtention de brevets de faible valeur. Mais l’autorisation marketing affecte-t-elle réellement si les compagnies pharmaceutiques poursuivent des brevets de suivi?
Dennis Byrski et Lucy Xiaolu Wang, CC By-NC-ND
Pour étudier comment les comportements de brevet changent après l’autorisation du marketing, nous avons spécifiquement utilisé des données du Bureau allemand des brevets et des marques et de la base de données statistique des brevets du Bureau des brevets européen. Les chercheurs juridiques et de l’innovation du monde entier considèrent souvent l’agence européenne comme l’étalon-or pour la qualité des brevets, et les chercheurs utilisent des brevets de médicament européens comme des références de haute qualité lors de l’évaluation des brevets américains de drogue.
En outre, les États-Unis ont vu quatre principaux affaires de la Cour suprême impliquant l’admissibilité aux brevets entre 2010 et 2014, dont deux axées sur le secteur pharmaceutique. Le cadre européen nous a permis d’étudier les changements dans le comportement de brevet en l’absence de modifications juridiques directes du système de brevets.
L’identification des brevets primaires n’est pas facile. Parce qu’ils ne sont souvent pas étiquetés dans des bases de données de brevets de médicament, les chercheurs doivent souvent examiner manuellement les longs textes de brevet pour les drogues américaines. Nous surmontons cette difficulté en suivant les certificats de protection supplémentaires accordés par le système européen de prolongation des termes de brevets. Ce système oblige les entreprises à préciser quel principal brevet de médicament à étendre après l’autorisation de marketing et avant l’expiration des brevets.
Nous avons constaté que la divulgation de l’art antérieur – comme les connaissances existantes à partir des données d’essai cliniques – pendant l’autorisation de marketing rend plus difficile l’obtention des brevets de faible valeur et de suivi par la suite. Cela s’est reflété par une forte baisse des auto-citations à partir de brevets ultérieurs pour ce médicament et d’autres brevets avec des cibles de maladie similaires.
En revanche, les auto-citations ultérieures des brevets de produit substantiel – tels que ceux des nouveaux dérivés de médicaments – et les brevets ciblant différentes zones de maladie se poursuivent à peu près au même rythme qu’avant l’autorisation de marketing.
Ces résultats suggèrent que la transparence dans le processus d’autorisation dissuade efficacement les entreprises d’obtenir des extensions de brevets à faible valeur sans décourager de nouvelles recherches et développement.
Surtout, nous avons vu des ajustements de brevet similaires parmi les concurrents, les collaborateurs et les fabricants génériques du titulaire des brevets. Ce modèle suggère que les changements dans les comportements de brevet peuvent ne pas être motivés par une recherche de profit réduite après l’approbation du médicament, car d’autres entreprises auraient une motivation plus élevée pour obtenir des brevets faibles liés après avoir vu le potentiel du marché d’un médicament. Une fois que les données des essais cliniques sont publiques, cela semble avoir un effet à l’échelle du système sur la réduction des brevets de faible valeur et de suivi, probablement tirés par une barre plus élevée pour la nouveauté.
Fait intéressant, nous n’avons pas vu de baisses similaires dans les dépôts de brevet après des étapes antérieures dans le processus de développement des médicaments, tels que la fin des essais cliniques de phase 2. Ces jalons fournissent des informations sur la qualité des médicaments mais impliquent moins de divulgation de données, ils sont donc moins susceptibles de fournir des arts antérieurs utilisables pour les examinateurs de brevets.
En d’autres termes, c’est la transparence clinique complète à l’autorisation de marketing qui fait une grande différence.
Ce que cela signifie pour les patients et les décideurs politiques
Qualités de brevet de médicament. Les brevets faibles peuvent augmenter les coûts des médicaments et retarder l’accès en bloquant la concurrence des génériques longtemps après que le marché a récompensé une entreprise pour sa principale innovation. Les résultats peuvent être coûteux pour les patients, les assureurs et les systèmes de santé publique, et il risque de diriger la R&D vers des ajustements marginaux au lieu de thérapies révolutionnaires.
Nos résultats suggèrent que l’intégration des informations réglementaires, y compris les données d’essais cliniques, dans les évaluations des brevets peut indirectement améliorer la qualité des brevets. Cela peut réduire le nombre de brevets de médicaments faibles déposés davantage pour des considérations stratégiques plutôt que d’améliorer la santé des patients.
Un meilleur alignement des brevets sur une véritable innovation n’est pas seulement une préoccupation juridique mais un impératif de santé publique. La transparence, associée à des systèmes d’examen plus intelligents, peut aider à augmenter la barre du développement des médicaments et à récompenser les types d’innovations qui améliorent vraiment la santé.
[ad_2]
Source link